General information
Globin chain involved
Status
Heterozygous
Migration zones
Migration positions
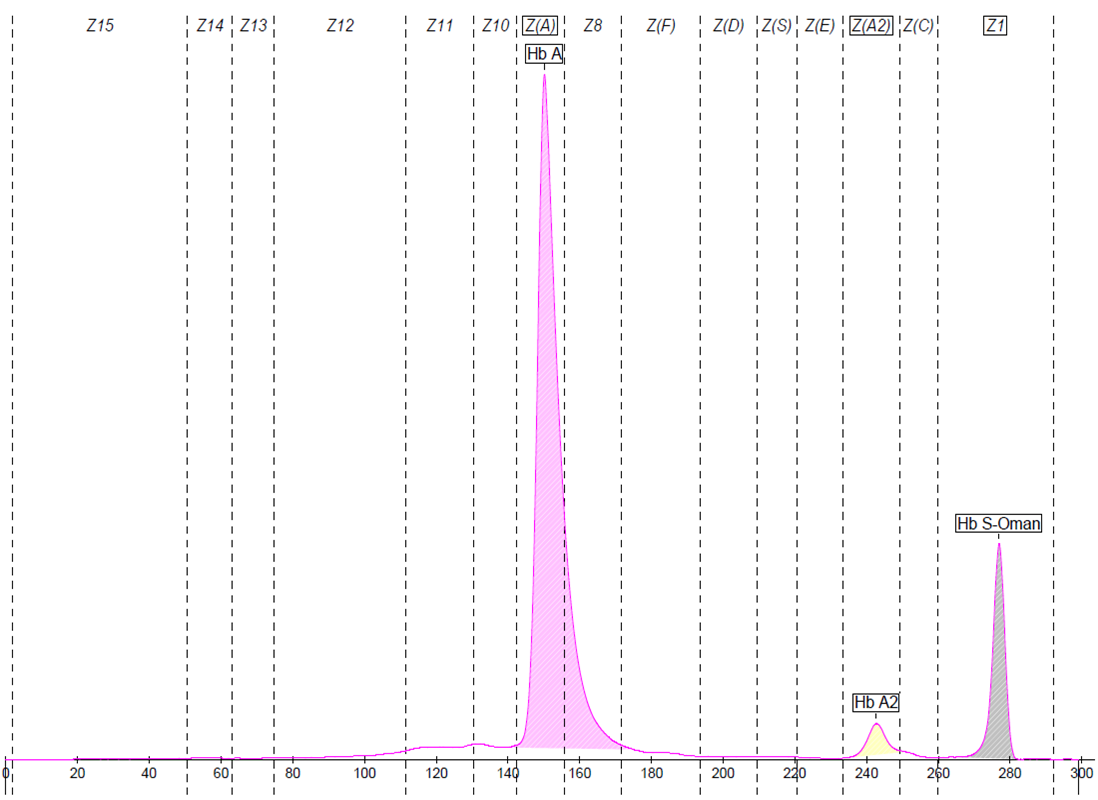
277
Sickle Cell Disease: Yes
Thalassemic variant: No
Capillary Electrophoresis
Fractions
Value %
Hb A
82.6
Hb A2
3.0
Hb S-Oman
14.4
Comments
A 14.4% expression of Hb S-Oman is consistent with the combination of this rare variant with homozygous alpha+-thalassemia (not studied during genotyping).
Mutation data
Heterozygous Hb S-Oman
Mutation
HGVS Nomenclature
Beta 6(A3) Glu>Val AND Beta 121(GH4) Glu>Lys
HBB:c.[20A>T;364G>A]
Hematological parameters
Name
Result
RBC Count
Low
Total Hemoglobin
Normal to low
MCV
Normal to low
MCH
Normal to low
Blood smear
Unique form of irreversibly sickled cell reminiscent of a “yarn and knitting needle” shape* (rare if the Hb S-Oman level is below 15%). Folded and target cells similar to those found in C/C, S/C, S/O-Arab, and homozygous Hb O-Arab. * Sometimes called Napoleon's hat x-shaped cells
Other analysis
No information
Comments on hematology
Reticulocytosis, hemolytic anemia.
Microcytosis linked to the concomitant presence of silent alpha-thalassemia or alpha-thalassemia trait
Clinical context
Clinical presentation
Clinical syndrome of Sickle Cell Anemia of moderate to high intensity in patients with 20% or more of Hb S-Oman, requiring frequent transfusions. No clinical syndrome for lower expression of Hb S-Oman (13-15%).
Clinical risk
Severe risk in combination with Hb S, Hb C, Hb E, beta-thalassemia, Hb Lepore
Variant information
Stability
Normal
Oxygen affinity
No data
Ethnicities in literature
Found in Middle East populations: met in a 24-year-old male from Oman, in a few other Omani families, and in a 3-year-old Omani girl carrying a compound heterozygosity of Hb S-Oman and Hb S
Comments on variant information
The rare variant Hb S-Oman results from a double mutation of the beta-chain (combination of Hb S and Hb O-Arab mutations, on the same beta gene) and is considerably less soluble than Hb S.
This variant has been found in combination with alpha+-thalassemia heterozygous (Hb S-Oman is expressed at 20%) and alpha+-thalassemia homozygous (Hb S-Oman is expressed at 14%).
Scientific Literature
Scientific references
- https://pubmed.ncbi.nlm.nih.gov/2930724/ Langdown J.V. et al., Br J Haematol. 1989 Mar;71(3):443-4.
- https://pubmed.ncbi.nlm.nih.gov/9834244/ Nagel R.L. et al., Blood. 1998 Dec 1;92(11):4375-82.
- https://pubmed.ncbi.nlm.nih.gov/11849204/ Al Jahdhamy R. et al., Br J Haematol. 2002 Mar;116(3):504.
- https://pubmed.ncbi.nlm.nih.gov/21748082/ Venugopal S. et al., Sultan Qaboos Univ Med J. 2008 Nov;8(3):344-6.
- https://pubmed.ncbi.nlm.nih.gov/28699687/ Al Balushi HWM et al., Br J Haematol. 2017 Oct;179(2):256-265.
Globin Chain involved
Status
The term "Double Heterozygous" refers to cases of heterozygosity on different globin chain types, while the term "Compound Heterozygous" refers to cases of heterozygosity on the same globin chain type.
For example, S/G-Pest is a Double Heterozygous case (beta and alpha-globin chains are mutated) and S/C is a Compound Heterozygous case (only beta-globin chains are mutated).
Migration zones
Migration positions
In some cases (homozygotes, combination of the variant with thalassemia, transfused patients, degraded samples or unstable variants), the variation in the migration position may be greater than +/- 1 point.
For profiles with thalassemia, only Hb A2 and Hb F peaks, if present, are listed with migration positions.
Sickle Cell Disease
Thalassemic variant
Capillary Electrophoresis
Variant information
Ethnicities are provided for informational purposes only and are based on scientific literature and conference posters.
A hemoglobin variant may therefore be present in populations of ethnic origins or countries not listed here.
Hematological Parameters