Hemoglobinopathies: Hemoglobin Variants and Thalassemias, description, pathophysiology and diagnosis
Preface by Dr. Cornelis L. Harteveld, PhD
Associate Professor, Dpt. Human and Clinical Genetics (Genome Diagnostics),
Hemoglobinopathies Reference Lab, LUMC – The Netherlands
1. Introduction
Severe hemoglobinopathies, read thalassemia major and Sickle Cell Disease, are the most common "autosomal recessive" hereditary disorders worldwide.
"Autosomal" means that males and females can be equally carriers or affected and "recessive" means that carriers of these disorders are generally healthy and therefore often unaware of their carrier status.
"Recessive" means also that children of parents who are both healthy carriers of the traits at risk (common traits at risk on Table 1) have 25% chance of being born severely affected, 50% chance of being a healthy carrier like their parents and 25% chance of not being carrier at all. If only one parent is a carrier the couple is not "at risk" but all children have 50% chance of being a healthy carrier and, on their turn, children may form couples at risk with partners who are also healthy carriers.
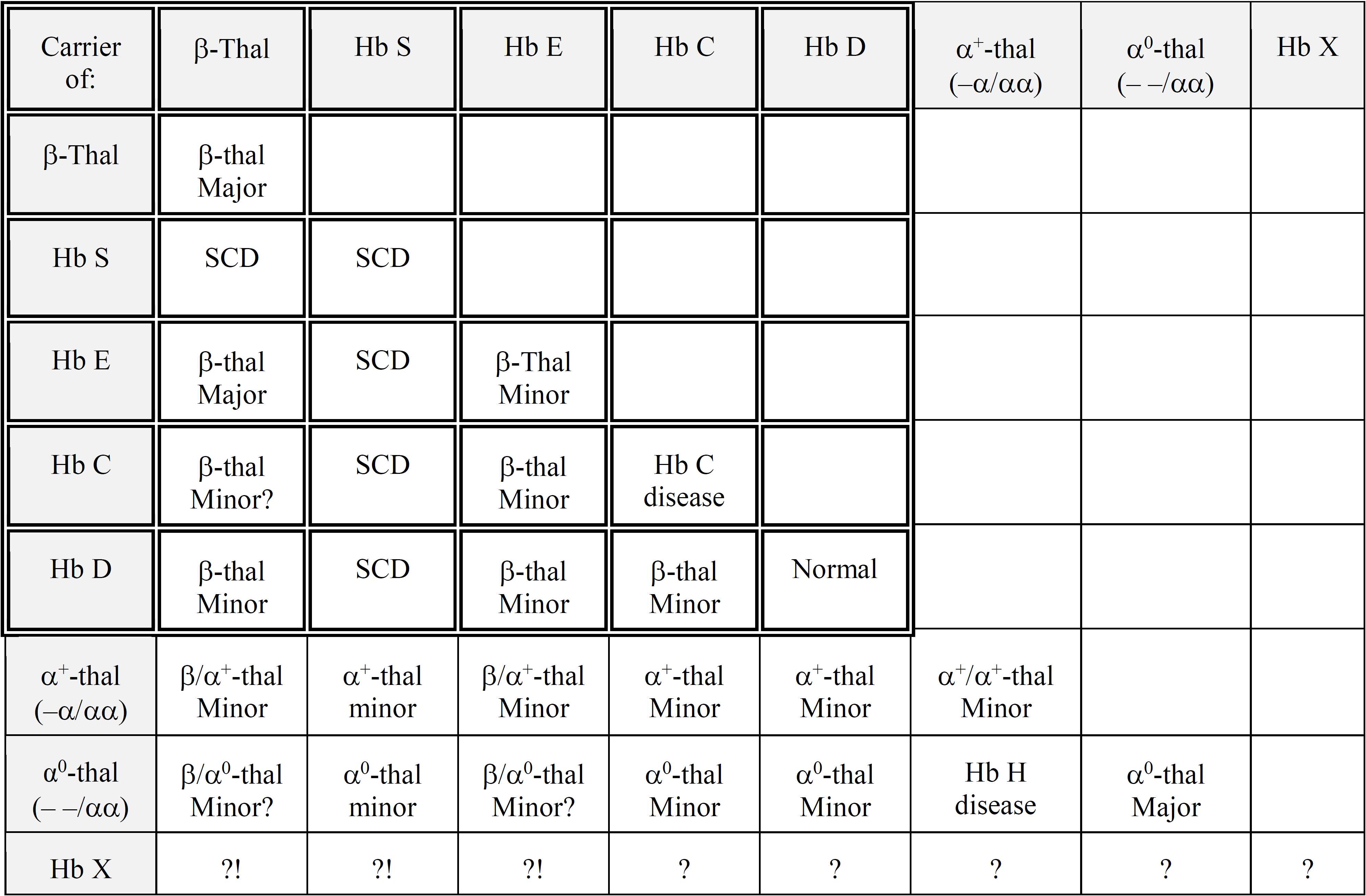
Table 1: Summary of the common "beta gene" (double-lined squares) and the common "alpha gene" traits and the genetic risk deriving from their combinations. For rare, unknown Hb X variants, "?" stands for unknown and "?!" for possible risk. Hb S: [β6(A3)Glu→Val, GAG>GTG; HBB: c.20A>T]; Hb E: [β26(B8)Glu→Lys, GAG>AAG; HBB: c.79G>A]; Hb C [β6(A3)Glu→Lys, GAG>AAG; HBB: c.19G>A]; Hb D: Hb D-Punjab [β121(GH4)Glu→Gln, GAA>CAA; HBB: c.364G>C] Hb E homozygosity is also known as Hb E disease, but is expressed as a beta-thal trait phenotype.
Geographical distribution
In the past, these disorders were mostly confined to the tropical and subtropical regions of the old world where, during the evolution of mankind, carriers were protected against premature death caused by malaria tropica.
Historical and more recent multiethnic migrations have spread hemoglobinopathies to virtually all corners of the world and in particular to immigration countries, where primary prevention has become a matter of great concern and carrier diagnostics are daily practice for clinical laboratories.
Diagnostics of Hemoglobinopathies is a Laboratory Matter
Severely affected children born from couples at risk for Beta-thalassemia Major (BTM) and Sickle Cell Disease (SCD) can be putatively diagnosed by newborn screening, and if screening is not available, they are in general clinically recognized between the first and the third year of life. However, the clinical diagnosis might become delayed especially in non-endemic countries and always needs, as newborn screening, confirmation at the laboratory level, first by basic family analysis (1) and then at the molecular level (2). DNA analysis confirms the basic laboratory diagnosis by providing the specific mutation and herewith the necessary information to allow reliable prognosis, a tailored treatment and the risk prediction required for an informed reproductive choice and eventually for prenatal diagnosis of a next pregnancy.
Diagnosis of Healthy Carriers. Why, When and How
Diagnosis is necessary to spare severe physical, psychological and economic burdens to parents, patients and public health by offering primary prevention for these severe heritable diseases. In addition, healthy carriers need to be reassured about their status. Their eventual chronic mild anemia, often mistaken for iron depletion, needs to be properly treated. As mentioned above, carriers can only be diagnosed by laboratory analysis that can be offered collectively during national screening campaigns or individually by regular diagnostics upon indication. Diagnosis should be offered, preferably before marriage or conception or in early pregnancy in order to allow an informed reproductive choice before an affected child is born. Carrier identification before marriage or pregnancy allows couples at risk to choose for more prevention alternatives such as adapting partner choice, remaining childless, considering adoption, considering egg or sperm donation or choosing for pre-implantation diagnostics, for regular or for non-invasive prenatal diagnosis (3).
Although it is the most practical alternative, screening in early pregnancy leaves prenatal diagnosis and medical abortion as the only prevention option in the case of an affected fetus. Newborn screening (NBS), applied mainly in non-endemic countries, is primarily meant for early diagnosis and treatment of the affected newborn and has a limited effect on primary prevention. Nevertheless, couples at risk identified because they had an affected child or a carrier, may get access to retrospective or prospective primary prevention for the next child if all carriers are reported.
Couples at risk who have a non-affected child are not identified by NBS until the next child is born affected or carrier (4).
Carriers can also be diagnosed individually by regular diagnostics upon indication. In case of carriers of thalassemia or thalassemic Hb variants, the indication will be chronic microcytic anemia not responding to iron medication. In case of unstable Hb variants or variants with abnormal oxygen affinity, the indication will be hemolysis, erythrocytosis or cyanosis. Nevertheless, considering that most carriers of traits associated with Sickle Cell Disease are usually not anemic and are asymptomatic, indications such as, origin from an endemic area, family history or routine control in early pregnancy need to be taken into consideration (1).
Purpose of this Atlas
The most useful tools a modern laboratory can use for basic hemoglobinopathy (HBP) diagnostics are the so called "dedicated" devices such as High-Performance Liquid Chromatography (HPLC) and Capillary Electrophoresis (CE) apparatuses (5). This author has published on several occasions different papers concerning the performances of these machines and has used for many years the Sebia Capillary Electrophoresis apparatus in particular, both as an independent device and as a complementary tool to HPLC. As an independent device, the CE is today the best automated alternative to classic electrophoresis. The instrument provides rapid and precise separation and measurement of all normal and of most abnormal Hb fractions in a matter of minutes. As complementary instruments, both CE and HPLC compensate for most of each other's imperfections (6-8).
The purpose of this Atlas is to share with laboratory technicians and physicians personal insights and examples of hemoglobin separation obtained with the SEBIA Capillary Electrophoresis device. Please read Atlas Presentation page for more details.
The previous version of the Hemoglobin Atlas presented examples subdivided into three categories. The first category included common defects at risk (shown in Table 1). These common variants are presented as they occur in the most frequent forms in daily practice, including the common combinations with alpha-thalassemia. The second category presented the less common variants associated or not with pathology and the third presented rare variants observed so far on CE. For this new version of the Hemoglobin Atlas, the subdivision into three categories has been removed, the occurrence of rare and less common hemoglobin variants is closely linked to the geographical origin of the patient and the location of the healthcare facility.
All cases reported in the Atlas have been confirmed either by protein or by DNA analysis in specialized laboratories. An identical separation pattern to one in this Atlas does not necessarily indicate the same mutation. However, the common mutations (Hb S, Hb C, and Hb E) can be considered identified at near 100% specificity because they are the most common and Hb S can be confirmed by the sickle test. This means that in the absence of contaminating blood transfusions all variants, except Hb S, need to be confirmed at the DNA level in case of potential couples at risk. For all other variants and for the rare ones in particular, the specificity can be estimated to be lower and DNA analysis is needed for confirmation.
2. Laboratory Diagnostics at Three Levels
Hemoglobinopathy diagnostics take place at 3 levels. The first, or basic diagnostic, is the kind that should allow most laboratories to work with dedicated devices and sufficient know how to provisionally diagnose patients and carriers of the common hemoglobinopathy conditions routinely. At this level the diagnosis is obtained basically by separation and measurement of the normal and abnormal hemoglobin fractions and by comparison with the basic hematological parameters. More details in reference (1).
The following level, or specialized diagnostics, is the kind that confirms the provisional conclusions reached by basic diagnostics, is mainly done at DNA level and should be performed in reference centers with sufficient experience in molecular diagnostics, possibly the same labs providing prenatal diagnosis. More details in reference (2).
The third level is the prenatal diagnosis itself that can be offered at 11 weeks of gestation performing DNA analysis on chorionic villi or on amniotic fluid at 14 weeks of gestation. Alternatively, non-invasive analysis of fetal DNA in maternal circulation can be offered in selected cases and at an earlier stage while Pre-implantation Genetic Diagnostics (PGD) can be offered under special circumstances before pregnancy (3). On behalf of contributors to the EMQN (European Molecular Genetics Quality Network) hemoglobinopathies best practice meeting held in Leiden in 2012 a group of international experts have defined a policy towards the molecular and hematologic methods used for carrier identification and prenatal diagnosis of the hemoglobinopathies. This has been published in the European Journal of Human Genetics and can be downloaded from the EMQN website (9).
3. The Molecular Background of Hemoglobinopathies
The Involved Genes
For a correct interpretation of the results at any level of diagnostics one must be aware of globin genes function and its expression.
The globin genes are clustered and located on different chromosomes (autosomes) of which we have a paternal and maternal copy. The β cluster is located on chromosome 11, contains one non-active pseudo gene (ψβ or HBBP1), one embryonic gene (ε, or HBE), two fetal genes (Gγ and Aγ, resp. HBG2 and HBG1) and two postnatal genes, the "low expression" δ (or HBD) and the "full expression" β (or HBB) gene. The two γ genes are active from late embryonic life until birth coding for fetal hemoglobin Hb F. The δ gene is still silent at birth and, in normal conditions, reaches 2.5 -3.5% expression within one year after birth. Since the β:δ expression rate is roughly 97:3, the most important genes in postnatal life are the two β genes, one from maternal and one from paternal origin.
The α cluster is located on chromosome 16 and contains 3 non-active pseudo genes, one embryonic (ζ or HBZ) gene and two α genes (α2 and α1, resp. HBA2 and HBA1) which are active during embryonic, fetal and postnatal life at the approximate rate of 2 to 1. The ζ gene codes with the ε gene for the first embryonic chains and Hb Gower 1. The two α genes contribute to the formation of the second embryonic Hb Gower 2 (α2/ε2), the fetal Hb F (α2/γ2), and both postnatal Hb A (α2/β2) and Hb A2 (α2/δ2). Noteworthy, four α genes (two from the maternal and two from the paternal chromosome 16) are contributing to the formation of Hb A and Hb A2 while only two β genes and two δ genes are providing the non-α counterpart to form these post-natal tetramers.
Normally there is a balanced expression, resulting in a balanced synthesis between alpha- and non-alpha-globin chains. However, variants in the globin genes resulting in a reduced or absent expression affect the synthesis ratio and may lead to thalassemia syndromes as explained below. (10).
Expression and/or Structure Defects
Hemoglobinopathies are caused by mutations on the globin genes. Some mutations impair the expression of the gene by reducing or totally eliminating the production of globin and herewith causing thalassemia. Other mutations may change the structure of the gene products generating abnormal globins and herewith hemoglobin variants. While structural defects are located on the coding regions of the genes (exons), expression defects can occur on any coding or regulating sequences of the gene and gene cluster.
The most common and clinically relevant expression defects are β- and α-thalassemias, caused by over 480- and 400-point mutations and deletions, respectively, in the corresponding globin genes (www.ITHAnet.eu). Virtually all thalassemia defects can be considered recessive and pathologically significant.
As mentioned above, the most common structural defects are Hb S, Hb C, Hb E and eventually Hb D-Punjab. These four variants are recessive and all may be associated, by gene combination, with Sickle Cell Disease or Beta-thalassemia Major (Table 1). However, over 1500 less common, rare or very rare Hb variants have been described during several decades and are registered on HbVar database(http://globin.bx.psu.edu/hbvar/menu.html) and ITHANET database (www.ithanet.eu). Of these, more than 70 can be considered more or less semi dominant, mainly because of their instability. Of the remaining variants, approximately one-third can be considered partially recessive, as they may lead to intermediate or severe phenotypes when co-inherited with β-thalassemia (in hemizygosity) or with Hb S.
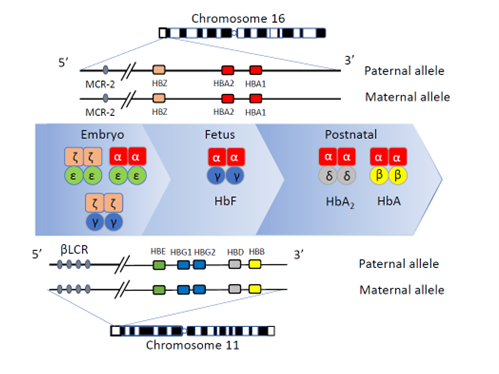
Figure 1: Schematic representation of the alpha-globin and beta-globin gene clusters. The clusters are located on chromosomes 16 and 11 respectively with the embryonic HBZ (ζ) and HBE (ε) genes, the fetal HBG1 and HBG2 (γ) genes, the embryonic, fetal and postnatal HBA1 and HBA2 (α) genes and the postnatal HBD and HBB (resp. δ and β) genes. The corresponding Hb tetramers Hb Gower 1 and 2, Hb Portland, Hb F, Hb A2 and Hb A are depicted from left to right with their different globin chains composition. Pseudo genes are not shown. Adapted from Farashi and Harteveld (11).
Recognizing the Abnormal Expression of the Genes Involved
Since we have only two HBB genes (figure 1), β-globin chain variants, if not unstable, will usually be synthesized in the carrier at about 40-50% rate unless the other HBB gene is thalassemic and has a reduced expression in which case the β variant will be more abundant. The common Hb E is one of the exceptions because the mutation also generates an alternative splice site and is therefore a "thalassemic variant". Hb E usually comes with ~25% expression in the plain heterozygote. Likewise, unstable (or other thalassemic) β variants, will show reduced synthesis and be eventually detectable as a trace or a smear of an abnormal Hb fraction or not be visible at all. Because of the 4 α genes (HBA1 and HBA2 on both alleles) (Figure 1) active in fetal and adult life at a different rate, α variants, if not unstable, will be synthesized at about 20-25% (α2) or at about 15-20% (α1). These percentages might become significantly increased (up to β variants levels) if an α-thalassemia defect is present on the opposite allele. Moreover, Hb A2 (α2/δ2) being also involved in the composition of α chain variants, these mutations will produce a second Hb A2 fraction (α*2/δ2), synthesized at the same proportional rate and positioned at about the same distance from the normal Hb A2 as the main α-globin variant has from Hb A. The estimation of the Hb A2 will then be the sum of the "two" Hb A2 fractions. If the minor HbA2 fraction overlaps with the HbA fraction, it may not be detectable.
Stable δ (HBD) gene variants may appear solely as a "second" Hb A2 fraction either faster or slower than the normal Hb A2 and be expressed at approximately equal rate. Also in these cases, the estimation of the Hb A2 will be the sum of the two fractions. Delta-thalassemia variants will be invisible and will lower by half the elevated Hb A2 value of the β-thalassemia carrier and compromise the diagnosis of β-thalassemia traits.
Variants of the γ genes (HBG1 and HBG2) may also produce Hb variants that are only visible at birth and will remain visible until the disappearance of Hb F. Due to the presence of 4 γ genes (figure 1), pathology associated with γ variants is rare and will disappear after the postnatal switch from Hb F to Hb A (12).
4. Provisional Diagnostics of the Common Traits
Taking into consideration that in hemoglobinopathy diagnostics exceptions are the rule, all high Hb A2 Beta-thalassemia carriers, with or without abnormally elevated Hb F levels, will be diagnosed by their elevated Hb A2 fraction (>4%) and hematological picture. The common Hb variants (Hb S, Hb C, Hb E and Hb D-Punjab) will be putatively identified in the presence or absence of Hb A, indicating the carrier or the homozygous or hemizygous state respectively. The first exception to eventually consider is that patients might have had a recent blood transfusion and present with altered patterns deriving from the mixture with donor blood.
As mentioned above, separation and measurement of the Hb fractions can identify the common conditions at high sensitivity. However, there are many rare variants that migrate or elute on CE and HPLC at the same position as the common fractions. Clinical chemists must therefore ensure that the presumptive diagnosis suggested by CE or HPLC is consistent with the complete blood count (CBC), iron status, red cell morphology (RCM), and the patient's ethnic background. These parameters and data should match the putative results and be compatible with the function of the protein (instability, hemolysis, oxygen affinity) and with the expected gene expression and clinical phenotype (1).
In case of discrepancies, the case should be further investigated at the DNA level, especially when genetic risk is involved.
In addition, the lab being probably the best expert on the matter, sufficient information should be provided to doctors and patients explaining the significance of the provisional results, giving advice to refer for carrier analysis either both parents or the partners or the children and grandchildren whenever a carrier is diagnosed in childhood, at young or at elderly age respectively and of course when a couple at risk is suspected.
5. Significance of Blood Counts, Iron Parameters and Red Cell Morphology in Hemoglobinopathy Diagnostics
Quite a few α and β Hb variants present with thalassemic phenotypes and with the hematological abnormalities of a thalassemia carrier. The carrier of an abnormal Hb with a thalassemic effect will present with a fraction at lower expression (i.e. Hb E), with some hemolysis if the fraction is partially unstable or with no fraction at all and with cellular inclusions if the instability is relevant. Moreover, common β variants like Hb S or Hb C, normally not present with thalassemic parameters, occur very often in association with alpha-thalassemia. Then the complete blood count (CBC) and red cell morphology (RCM) will reflect the thalassemia conditions, which will also reduce the expression of the Hb variant progressively.
The classic β-thalassemic phenotype presents microcytic hypochromic anemia. Ferritin will generally be elevated and, if folic acid is not depleted, RBC counts will be elevated too. RCM will show the characteristic target cells, basophilic stippling and anisopoikylocytosis of thalassemia condition. Common β variants like Hb S are often inherited together with β-thalassemia. This condition called hemizygosity (Hb S/β-thal) presents clinical features like SCD and is not distinguishable from homozygosity (Hb S/S) on CE or HPLC. However, in these cases the CBC will show microcytic anemia and the Hb A2 level will be reliably elevated on CE (not on HPLC).
As with β-thalassemia traits, most α-thalassemia carriers will present with normal CE or HPLC patterns in postnatal life. Only Hb H disease will show variable amounts of (unstable) Hb H (β4). Conversely, all α- thalassemia carriers will show variable amounts of Hb Bart's (γ4) at birth. As in β-thalassemia traits, α-thalassemia carriers are usually microcytic. However, because of the presence of 2 α genes on chromosome 16 (4 in total) the pathology of α- thalassemia comes in 4 different forms.
Carriers of a single α gene defect (-α/αα, heterozygous α+) are usually asymptomatic but can be slightly anemic, microcytic and hypochromic. Carriers of two α gene defects, either in cis (--/αα, heterozygous α0) or in trans (-α/-α, homozygous α+) are always slightly anemic, microcytic and hypochromic. In both single and double α gene defects, RCM will be moderately abnormal in adults with microcytes, target cells, basophilic stippling and anisopoikylocytosis. In the heterozygous α0-thalassemia carrier, stained with Brilliant Cresyl Blue (BCB) will show a few rare but typical Hb H inclusion bodies. In Hb H disease (--/-α), microcytic and hypochromic anemia vary from intermediate to severe, hemolysis will be present, as well as hepatosplenomegaly and skeletal abnormalities. These patients may need blood transfusion. RCM will be strongly altered with microcytes, target cells, basophilic stippling and anisopoikylocytosis and BCB staining will show many red cells with Hb H inclusion bodies. Total absence of α gene expression (--/--, homozygous α0) leads to the lethal Hb Bart's Hydrops Fetalis Syndrome with a dramatic CBC and red cell morphology. Finally, alleles with a single α gene deleted (-α/αα) are often less pathogenic than alleles with a single point mutation (αTα/αα). In some cases, this is due to the nature of the mutation, as for instance the unstable Hb Constant Spring, common in population from Southeast Asia, affecting the dominantly expressed α2-gene and causing more severe Hb H phenotypes when combined with a deletion type α0-thalassemia, than point mutations affecting the expression of the α1-gene. CBC, iron and RCM data are summarized in table 2.
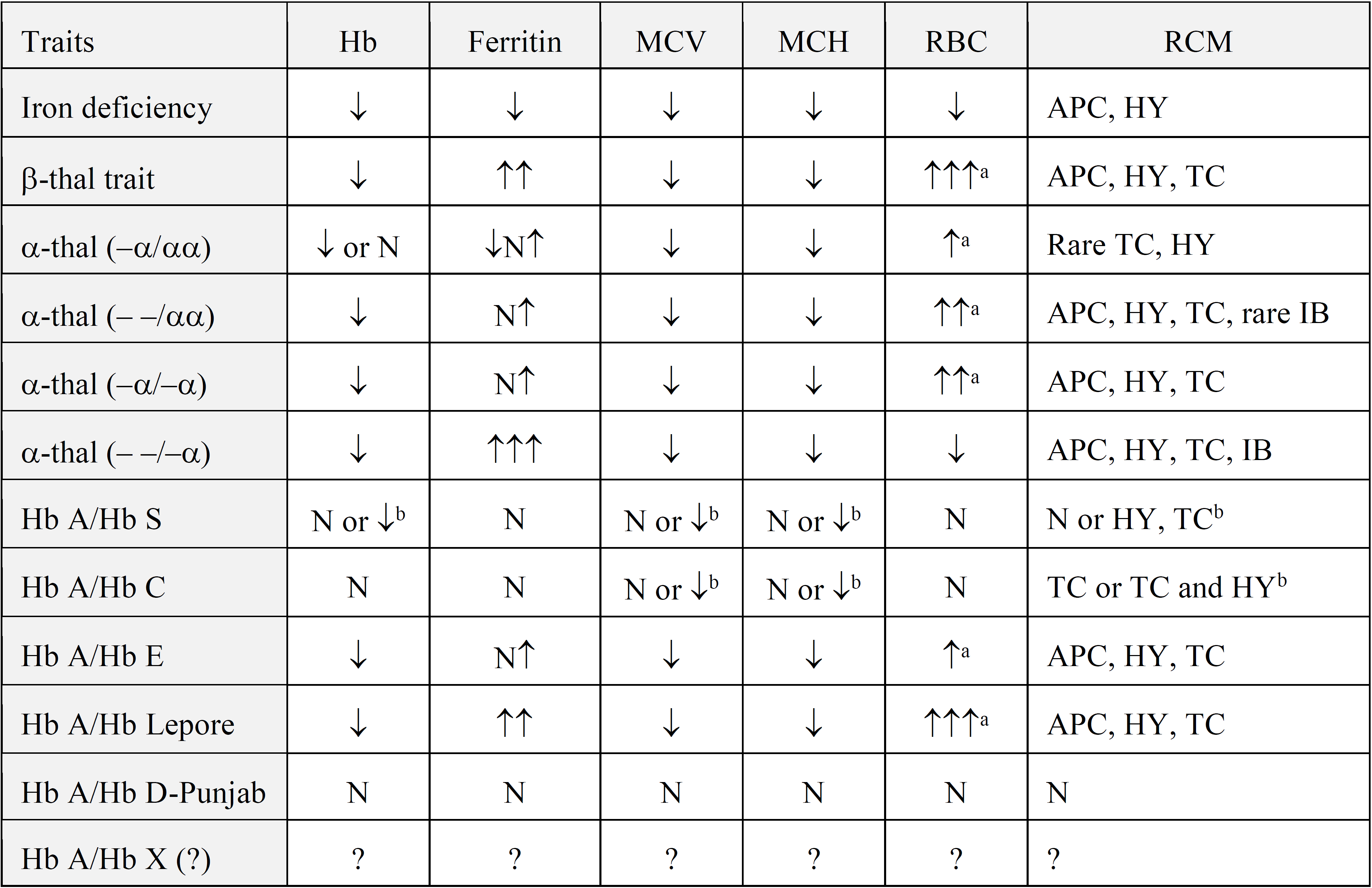
Table 2: Summary of the most significant values as generally observed from complete blood count, ferritin and red cell morphology (RCM) tests in the common traits at risk for intermediate or severe conditions. MCV= mean corpuscular volume; MCH= mean corpuscular Hb; N= normal; APC = anisopoikylocytosis; HY= hypochromia; TC= target cells; IB= inclusion bodies; a = unless folic acid depleted; b = if associated with alpha- thalassemia. Adapted from Giordano 2012 (1)
6. Significance of Normal and Abnormal Hemoglobin Fractions in the Common Conditions in More Details
β-Thalassemia
An adult β-thalassemia carrier will usually show 4% or more Hb A2 and eventually some Hb F (up to 7-8%) without further abnormalities. Then, if matching with the expected CBC parameters and if no HIV is involved, the provisional diagnosis carrier of β-thalassemia is done. Some β-thalassemia mutations come with near normal Hb A2 values or even with normal levels. Excluding a β-thalassemia trait just because of a normal Hb A2 level (2.5-3.5% values obtained by HPLC or 2.2-3.2% values obtained by CE) and trusting on the presumed precision of the measurement is very imprudent when CBC indicates microcytosis in absence of iron depletion. Then a β-thalassemia with borderline Hb A2, an α-thalassemia, a co-existing δ-thalassemia or a large beta deletion defect could be present (13) and molecular analysis is needed.
Over and Underestimation of Hb A2
Dedicated devices may overestimate or underestimate the Hb A2 levels in the presence of Hb variants. However, measurement of Hb A2 in the presence of Hb A and any β variant is obsolete. The presence of both Hb A and the β variant (in non-transfused patient) means that both β genes are expressed and that there is no need to measure Hb A2 to establish the presence of a non-expressed β-thalassemia gene.
α-Thalassemia
As already mentioned, adult α-thalassemia carriers will show no specific characteristics on electrophoresis, HPLC or CE except for a marginal reduction in Hb A2 expression (13). The only exception is Hb H disease which will usually present with an unstable and quickly disappearing Hb H (β4) fraction migrating in Z15 in CE. When suspecting α-thalassemia because of the CBC parameters in absence of iron depletion and low Hb A2, then molecular analysis is needed to detect the common deletions or point mutations (1). On the other hand, α-thalassemia can easily be detected at birth by the presence of Hb Bart's (γ4) (14). With levels of 1.8% ± 0.5% Hb Bart's is usually measured with a single non-active α gene; 9.2% ± 1.1% when 2 α genes are not active (either in cis --/αα or trans -α/-α) while in Hb H disease (-α/--), Hb Bart's levels will reach 24% to 25%, using HPLC. Values obtained by CE might be different (15). In case of the lethal Hydrops Fetalis syndrome (--/--), no Hb F is present in the fetus or at birth but traces of Hb H (β4); Hb Bart's (γ4); (ε4), Hb Gower 1 (ζ2ε2) and the embryonic Hb Portland (ζ2γ2) (16).
High Hb F in Adults
The normal amount of Hb F two years after birth is usually less than 0.5% in CE. All elevations of Hb F in adults have a reason. Modest elevations (2-2.5%) can be artifacts if measured on HPLC due to overlapping with one of the glycated Hb A fraction. Substantial elevations can be associated with bone marrow malignancies, aplastic anemia, Fanconi anemia, hereditary persistence of fetal hemoglobin (HPFH), point mutations on the promoters of the Gγ or Aγ gene, erythropoietic stress, treatment with particular cytotoxic agents (e.g. hydroxyurea) or pregnancy (17).
In carriers of β-thalassemia, the Hb F level can be normal or elevated up to 7-8% depending on the erythropoietic stress and the presence of frequent mutations on the Gγ (HBG2) gene promoter such as the Xmn-I polymorphism (HBG2 c.-211C>T). More elevated Hb F values associated with low Hb A2 levels may indicate δβ-thalassemia deletion defects. As Hb F cells are usually bigger than the Hb A cells, microcytosis can be masked in δβ-thalassemia with high Hb F levels.
Sickle Cell Trait
An abnormal adult pattern with ~40% Hb S over 60% Hb A and a normal Hb A2 and Hb F levels matching with the expected normal CBC parameters will indicate a simple carrier of Hb S. However, many rare abnormal hemoglobins, either known or yet unknown, migrate or elute on the same position (18). Moreover, a reduced level of Hb S may indicate a non-Hb S variant or co-inheritance of α-thalassemia, the last being very common.
Hb S can also become strongly reduced in carriers with profound iron depletion (19). The provisional diagnosis "Hb S carrier" obtained by traditional or Capillary Electrophoresis or by HPLC is quite robust but needs confirmation. The confirmation of Hb S is easy without molecular analysis using one of the commercial solubility tests or the reliable and simple sickle test as previously described (20). Alternatively, molecular diagnostics will be required when doing analysis for a possible couple at risk.
Other common "Sickle Cell Trait" variants
Hb C and Hb D-Punjab are silent traits causing only asymptomatic or mild conditions in homozygous forms or in combination with β-thalassemia respectively. However, both traits cause SCD when co-inherited with Hb S and have therefore the genetic effect of Sickle Cell Trait conditions. The same is true for virtually all β-thalassemia traits causing SCD in combination with Hb S and for Hb E being a mild condition in homozygous state but causing severe (transfusion dependent) beta-Thalassemia Intermedia in combination with β-thalassemia and SCD in combination with Hb S.
Hb C, Hb E and Hb D-Punjab are easily but putatively identified at near 100% sensitivity and over 90% specificity on CE not only because of the discriminating capacity of the machine but mainly because these variants are like Hb S the most common worldwide.
Hb C and Hb E migrate on the Hb A2 position on alkaline electrophoresis but are distinguishable because of their intensity. Hb C, Hb E and Hb D-Punjab have specific positions on CE, and they all separate from Hb A2 on CE. One may wonder what can be the use of measuring Hb A2 in the presence of Hb A and of any other beta variant when the presence of two β gene products tells us that both HBB genes are active and that consequently no β-thalassemia defect can be present.
Take Home Message
Up to date more than 2400 DNA variants causing Hb variants or thalassemia have been described in the ITHAnet and HbVar hemoglobinopathy databases, of which more than half are structural variants. Most of these are either rare or very rare and need to be characterized after HPLC or CE detection. Even though the majority of these rare Hb variants are recessive or non-pathological, some will be semi-dominant (the unstable and hyper-unstable hemoglobin variants and those with changes in oxygen affinity). The pathological ones with a semi-dominant phenotype usually do not escape clinical observation but might escape detection on HPLC or CE because of increased instability. Then, even with a non-informative separation one should trust upon the classic Red Blood Cell Count and go straight for DNA sequencing because these invisible variants may cause severe conditions in combination with α, β-thalassemia or Hb S. Moreover, a few rare traits with double mutations (Hb S, Hb C or Hb E with a second structural change) have been described. These variants elute on different zones than Hb S, Hb C or Hb E but may cause SCD or severe thalassemia in combination with Hb S, Hb C, Hb D-Punjab, Hb E or β-thalassemia (1).
Cornelis L. Harteveld, PhD
Associate Professor, Clinical biochemical molecular geneticist
Department of Human and Clinical Genetics / Genome Diagnostics
Hemoglobinopathy Expert Center
Leiden University Medical Center (LUMC)
Leiden, The Netherlands
7. References
1. Giordano PC. Strategies for basic laboratory diagnostics of the hemoglobinopathies in multi-ethnic societies: interpretation of results and pitfalls. Int J Lab Hematol. 2013 Oct;35(5):465-79. doi: 10.1111/ijlh.12037. Epub 2012 Dec 7
2. Harteveld CL. Laboratory diagnostics of hemoglobinopathies; State of the art and new developments in molecular diagnostics for hemoglobinopathies in multi-ethnic societies. Int J Lab Hematol. 2013 May 31. doi: 10.1111/ijlh.12108. [Epub ahead of print]
3. Traeger-Synodinos J. Preimplantation genetic diagnostics, an alternative to prenatal diagnosis of the hemoglobinopathies. Int J Lab Hematol. 2013 Dec;35(6):571–579. doi: 10.1111/ijlh.12086.
4. Giordano PC. Prospective and retrospective primary prevention of hemoglobinopathies in multiethnic societies.Clin Biochem. 2009 Dec;42(18):1757-66.
5. Van Delft P, Lenters E, Bakker-Verweij M, de KorteM, Baylan U, Harteveld C.L. and Giordano PC. Evaluating five dedicated automatic devices for hemoglobinopathy diagnostics in multiethnic populations. Int J Lab Hematol. 2009 Oct;31(5):484-95.
6. Giordano PC. Newborn screening for hemoglobinopathies using capillary electrophoresis. Methods Mol Biol. 2013;919:131-45.
7. Mantikou E, Harteveld CL, Giordano PC. Newborn screening for hemoglobinopathies using capillary electrophoresis technology: Testing the Capillarys Neonat Fast Hb device. Clin Biochem. 2010 Nov;43(16-17):1345- 50.
8. Mantikou E, Arkesteijn SG, Beckhoven van JM, Kerkhoffs JL, Harteveld CL, Giordano PC. A brief review on newborn screening methods for hemoglobinopathies and preliminary results selecting beta thalassemia carriers at birth by quantitative estimation of the HbA fraction. Clin Biochem. 2009 Dec;42(18):1780-5.
9. Traeger-Synodinos J. , Cornelis L Harteveld CL, Old JM, Petrou M, Galanello R, Giordano PC, Angastioniotis M, De la Salle B, Henderson S, May A. EMQN Best Practice Guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. Eur J Hum Genet. 2015 Apr;23(4):426-37.
10. Weatherall DJ, Clegg JB. The thalassemia syndromes, 4th edition. 2001.
11. Farashi S, Harteveld CL. Molecular basis of α-thalassemia. Blood Cells Mol Dis. 2018. May;70:43-53.
12. Zimmermann-Baer U, Capalo R, Dutly F, Saller E, Troxler H, Kohler M, Frischknecht H. Neonatal cyanosis due to a new (G)γ-globin variant causing low oxygen affinity: Hb F-Sarajevo (G)γ102(G4) Asn→ Thr AAC>ACC]. Hemoglobin. 2012;36(2):109-13.
13. Stephens AD, Angastiniotis M, Baysal E, Chan V, Fucharoen S, Giordano PC, Hoyer JD, Mosca A, Wild B; International Council for the Standardisation of Haematology (ICSH). ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol. 2012 Feb;34(1):1-13.
14. Papassotiriou I, Traeger-Synodinos J, Vlachou C, Karagiorga M, Metaxotou A, Kanavakis E, Stamoulakatou A. Rapid and accurate quantitation of Hb Bart's and Hb H using weak cation exchange high performance liquid chromatography: correlation with the alpha-thalassemia genotype. Hemoglobin. 1999 Aug;23(3):203-11.
15. Winichagoon P, Svasti S, Munkongdee T, Chaiya W, Boonmongkol P, Chantrakul N, Fucharoen S. Rapid diagnosis of thalassemias and other hemoglobinopathies by capillary electrophoresis system. Translational Research 2008; 152:178-184.
16. Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010 May 28;5:13. Review.
17. Stephens AD, Angastiniotis M, Baysal E, Chan V, Davis B, Fucharoen S, Giordano PC, Hoyer JD,Mosca A, Wild B; International Council for The Standardisation of Haematology (ICSH). ICSH recommendations for the measurement of haemoglobin F. Int J Lab Hematol. 2012 Feb;34(1):14-20.
18. Szuberski J, Oliveira JL, Hoyer JD. A comprehensive analysis of hemoglobin variants by high-performance liquid chromatography (HPLC). Int J Lab Hematol. 2012 Jun 20. [Epub ahead of print].
19. Giordano PC, Huisman W, Harteveld CL. Iron depletion: an ameliorating factor for Sickle Cell Disease? ISRN Hematol. 2011;2011:473152. Epub 2011 Jul 5.
20. Harteveld CL, Ponjee G, Bakker-Verweij M, Arkesteijn SG, Phylipsen M, Giordano PC. Hb Haaglanden: a new nonsickling β7Glu>Val variant. Consequences for basic diagnostics, screening, and risk assessment when dealing with HbS-like variants.Int J Lab Hematol. 2012 Oct;34(5):551-5.