General information
Globin chain involved
Status
Homozygous
Migration zones
Migration positions
252
Sickle Cell Disease: No
Thalassemic variant: No
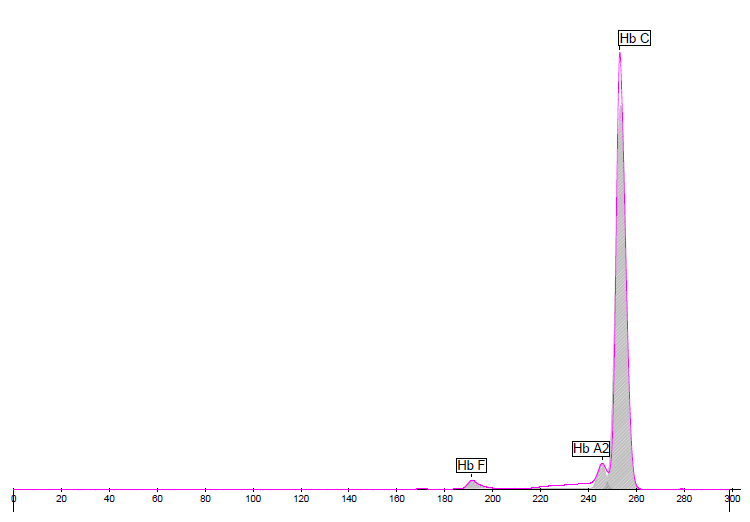
Capillary Electrophoresis
Fractions
Value %
Hb F
2.1
Hb A2
3.3
Hb C
94.6
Comments
Due to the homozygosity of the Hb C variant, the Hb A2 fraction is sometimes not perfectly quantified, or even identified as a single peak (both Hb A2 and Hb C peaks are then quantified jointly). If the Hb F fraction is very low, the profiles do not show any identified fraction as in the present case (no zone).
Mutation data
Homozygous Hb C
Mutation
HGVS Nomenclature
Beta 6 (A3) Glu>Lys
HBB:c.19G>A
Hematological parameters
Name
Result
RBC Count
Normal
Total Hemoglobin
Normal to low
MCV
Low
MCH
Low
Blood smear
Target cells, irregularly contracted cells, crystals
Other analysis
Reticulocytes, elevated MCHC, elevated viscosity, low hematocrit-to-blood viscosity ratio (HVR)
Comments on hematology
Microcytic, aniso-poikilocytosis, reticulocytosis
Clinical context
Clinical presentation
Mild hemolytic anemia, splenomegaly is very common, occasional hepatomegaly. No painful vaso-occlusive crisis or acute chest syndrome
Clinical risk
Intermediate to severe risk in combination with Hb S (results in Sickle Cell Disease).
Mild to intermediate in combination with beta-thalassemia, Hb Lepore and other less common hemoglobin variants with a thalassemic phenotype.
Variant information
Stability
Normal
Oxygen affinity
Normal
Ethnicities in literature
Found in Black African populations: very common in Black families, mainly of West African origin, but also reported in many other ethnic groups.
Comments on variant information
The very common Hb C variant has been found in combination with a large number of other hemoglobin variants and thalassemias, including Hb S, Hb E, Hb O-Arab, Hb Hope, Hb Korle-Bu, etc., as well as in the homozygous state (called Hb C disease). The presence of Hb C makes the blood more viscous and reduces red blood cell lifespan.
Scientific Literature
Scientific references
- https://pubmed.ncbi.nlm.nih.gov/13108995/ Ranney H.M. et al., J Clin Invest. 1953 Dec;32(12):1277-84.
- https://pubmed.ncbi.nlm.nih.gov/7229029/ Fabry M.E. et al., J Clin Invest. 1981 May;67(5):1284-91.
- https://pubmed.ncbi.nlm.nih.gov/23297836/ Cook C.M. et al., Hemoglobin. 2013;37(1):16-25.
- https://pubmed.ncbi.nlm.nih.gov/25712976/ Mangano V.D. et al., J Infect Dis. 2015 Aug 15;212(4):626-34
- https://pubmed.ncbi.nlm.nih.gov/25335812/ Lemonne N. et al., Clin Hemorheol Microcirc. 2016;61(4):571-7.
- https://pubmed.ncbi.nlm.nih.gov/38372896/ Aljabry M. et al., J Epidemiol Glob Health. 2024 Jun;14(2):298-303.
- https://pubmed.ncbi.nlm.nih.gov/40196068/ Regragui I. et al., Cureus. 2025 Mar 8;17(3):e80255.
Globin Chain involved
Status
The term "Double Heterozygous" refers to cases of heterozygosity on different globin chain types, while the term "Compound Heterozygous" refers to cases of heterozygosity on the same globin chain type.
For example, S/G-Pest is a Double Heterozygous case (beta and alpha-globin chains are mutated) and S/C is a Compound Heterozygous case (only beta-globin chains are mutated).
Migration zones
Migration positions
In some cases (homozygotes, combination of the variant with thalassemia, transfused patients, degraded samples or unstable variants), the variation in the migration position may be greater than +/- 1 point.
For profiles with thalassemia, only Hb A2 and Hb F peaks, if present, are listed with migration positions.
Sickle Cell Disease
Thalassemic variant
Capillary Electrophoresis
Variant information
Ethnicities are provided for informational purposes only and are based on scientific literature and conference posters.
A hemoglobin variant may therefore be present in populations of ethnic origins or countries not listed here.
Hematological Parameters